


01 Sep Not all mutations are equal
In a previous blog post (see July), we described the task of sequence alignments as trying to match the similar regions between genes of different organisms to understand better what corresponds to what, and characterize how they mutated away from their common ancestors.
We also discussed the issue of not having access to a “perfect” solution when it comes to alignments. We would like to take more time in this blog post to delve in the details of why that is the case.
We have access to many examples of “good” alignments. We know these are good because the frequency of mutations (the regions that don’t align perfectly) is consistent with what we know about the evolutionary distance between the two species. This evolutionary distance is, intuitively, a measure of how long ago the common ancestor of the two species was around.
For instance, humans and chimpanzees are quite closely related, as they would have diverged from their common ancestors around 7 to 10 million years ago (White et al, 2009). However, the common ancestor of humans and bats could have lived as long ago as 65 million years ago (or a few hundred thousand years after the extinction of the dinosaurs) (O’Leary, M. A. et al. Science 339, 662–667 (2013)). This means we would expect, when comparing genes between humans and chimpanzees, a lot more similarity than when comparing humans and bats.
We can also take this intuition further, quantitatively, by comparing closely related species to one another and to humans. The scientific study of evolutionary relationships between species is named phylogeny, and the most observable result of phylogeny research is the drawing of phylogenetic trees, which map evolution of related species from a common ancestor to separate species today.
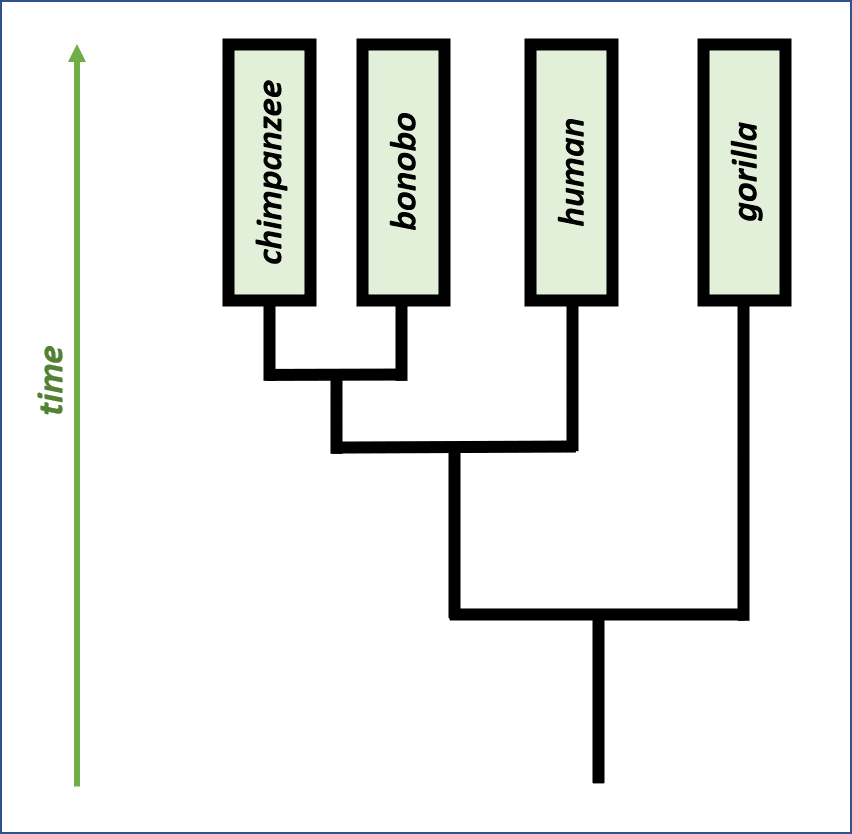
Phylogeny can be used to validate DNA alignments since it provides an alternate source of knowledge about how “similar” the two sequences should be. Unfortunately, we have limited phylogenetic information when it comes to the bacteria featured in this project; one of the objectives of Borderlands Science and its alignments is to learn more about the evolutionary relationships between bacteria.
Another important consideration is that variations observed in the genes of modern species are not random. A mutationcan occur following an error made during replication (when the organism makes a copy of the gene). This can happen pretty much anywhere in someone’s DNA sequence, and will affect the function associated with that gene.
You can see DNA like a “recipe” to make some organism. Each gene would be an ingredient and details on how to prepare it, and a mutation would be an error in these notes which could lead you to use the wrong ingredient, or prepare it incorrectly. However, many ingredients are absolutely essential for the recipe to work. If there is a mutation in them, the recipe will simply not work, and the organism will not survive, or not be able to reproduce. In other words, there will be no trace that it ever existed.
And that is the key: the mutations that we can observe today are the mutations that were not “lethal”. The regions of DNA that cannot be changed without causing death will remain identical among every living individual of a species; there are DNA sequences that every single live human have in common.
This principle can be leveraged when building and evaluating alignments; when looking at homolog genes (genes that are “recipe instructions” for the same ingredients), there will be regions that are perfectly identical among every living individual of that species, and the first step of the work would be to use those regions to start the alignment, and then look at how non-ubiquitous regions differ.
This means that, when it comes to the tasks shown in borderlands science puzzles, some regions are represented more than others, and user solutions will differ from computer solutions more markedly in some regions that others, because there are areas for which everyone will agree on the alignment (because everything is pretty much identical across all species). This leads to a very heterogeneous dataset, and one of the first problems to tackle as we move forward with producing new puzzles is to identify the regions that have the highest divergence between players and computers, as those are the ones that are most likely to include new information.
Indeed, it is impossible to learn about evolutionary divergence from looking at identical sequences between two species; all the information is about how, and how much they differ.
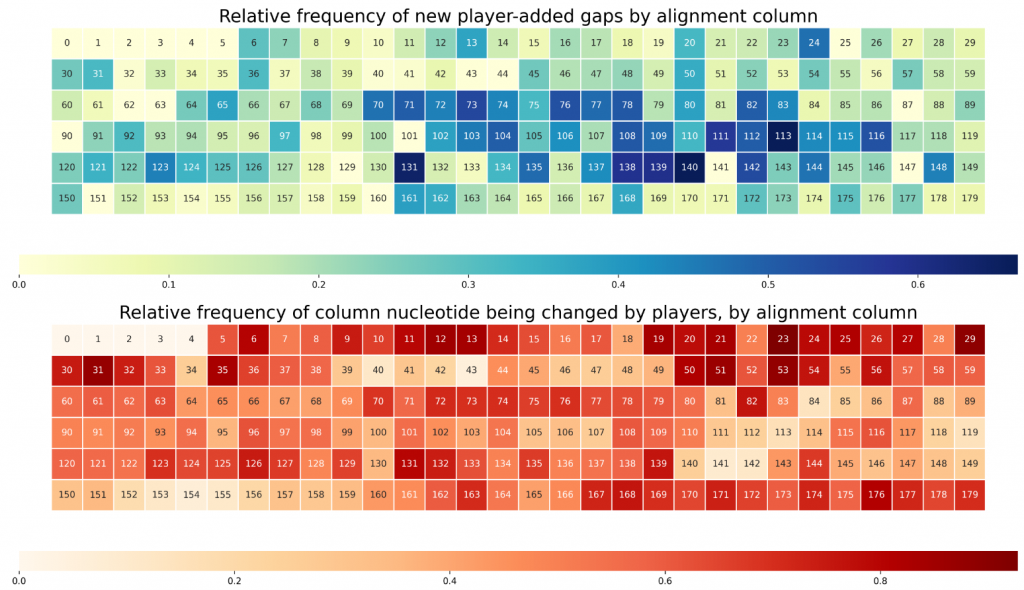
We can characterize this type of information with heatmaps.
The first heatmap includes information about the frequency of the addition of gaps by players, by column. In other words, it shows where, in the sequence, the players tend to add extra gaps (or identify extra insertion/deletion mutations).
The second heatmap encodes similar information, except instead of focusing on gaps, it focuses on substitutions.
When looking at both, we can observe that some regions are rich both in substitutions and in insertions/deletions (70 to 80, for example). However, some regions are very rich in one but not particularly rich in the other (10-15). One of our scientific goals will be to understand what this means in terms of the evolutionary relationship between gut bacterial species.